Questões
- Definir e diferenciar tumores sólidos e não sólidos.
- Descrever a origem e o desenvolvimento da linhagem hematopoiética.
- Descrever a fisiopatologia das leucemias.
- Explicar os sinais e sintomas, correlacionando-os com a patogênese e evolução das leucemias (destacando sua eventual semelhança com outras doenças menos graves).
- Citar as diversas leucemias, suas formas de apresentação e sua epidemiologia, relacionando: tipo de leucemia, faixa etária e prognóstico.
- Descrever os exames utilizados para o diagnóstico e estadiamento de leucemias.
- Identificar as principais formas e fases do tratamento das leucemias e suas indicações.
- Discutir a importância da equipe multidisciplinar para acompanhamento de pacientes e seus familiares.
- Identificar o papel das entidades de apoio ao paciente com câncer e familiares.
Respostas
-
Definir e diferenciar tumores sólidos e não sólidos.
A progressão do câncer (ou neoplasia maligna) é um processo complexo envolvendo várias escalas temporais e espaciais, um grande número de rotas metabólicas e diferentes tipos celulares. Em função disso, o termo “câncer” designa um conjunto altamente heterogêneo de doenças que podem afetar qualquer parte do corpo e que têm em comum o crescimento celular desordenado, a invasão de tecidos adjacentes e a formação de metástase.
Entretanto, poucos tipos de neoplasias respondem pela maior parte dos casos de cânceres fatais, sendo a grande maioria formadora de tumores sólidos. No Brasil cerca de 90,0% do total de mortes por câncer em ambos os sexos está associada ao desenvolvimento de neoplasias sólidas, enquanto que neoplasias de origem hematológica (e.g. leucemias e linfomas) respondem por menos de 7,0% dos casos.
As neoplasias de origem hematológica têm sido historicamente tratadas e classificadas separadamente das demais neoplasias, as quais são coletivamente referidas como neoplasias sólidas (ou tumores sólidos). Diferentemente das neoplasias hematológicas, as neoplasias sólidas formam massas tumorais que em geral ficam restritas às barreiras teciduais por um longo período durante os estágios iniciais da progressão tumoral até adquirirem características invasivas quando então conseguem irromper o compartimento de origem e colonizar novos sítios. Além disso, no decorrer da progressão tumoral, as neoplasias sólidas adquirem um número muito maior de alterações citogenéticas e moleculares. Como resultado, nos estágios mais avançados do desenvolvimento elas exibem acentuada heterogeneidade intratumoral – com formação de múltiplos clones aberrantes.
Carcinomas são originários de células do tecido epitelial de algum órgão. Se origina quando uma célula normal sofre mutação maligna e se multiplica. É um dos tipos mais comuns, podendo surgir em todos os órgãos. Há diversos tipos diferentes, entre eles o adenocarcinoma, que é quando a doença surge de um tecido epitelial que contém glândulas.
Sarcomas são neoplasias malignas que se desenvolvem a partir de tecidos ósseos e musculares, podendo estar localizados em diferentes partes do corpo, por exemplo, na musculatura da cabeça e pescoço ou das pernas. Os sarcomas dos tecidos moles e os osteossarcomas, que são os que atingemos ossos, estão entre os mais comuns. São os que costumam fazer mais metástase, que é a disseminação para outros órgãos.
Leucemias são tipos de câncer que inicialmente acometem a medula óssea, em que as células anormais, que sofreram mutação, se multiplicam e substituem as células normais, afetando a produção de glóbulos vermelhos, brancos e plaquetas no sangue. Há quatro tipos de leucemia: aguda, crônica, linfoide e mieloide.
Linfomas são cânceres que se desenvolvem nos gânglios linfáticos, que compõem o sistema linfático, distribuído em todo o corpo. Surge quando um linfócito, que é um tipo de glóbulo branco, torna-se célula maligna e cresce descontroladamente. Podem afetar pessoas de todas as idades, mas são mais comuns na fase adulta. Os dois tipos de linfomas são de Hodgkin e Não Hodgkin. Só deste segundo grupo, há ainda 20 subtipos.
Referência: ESTUDO DA DIVERSIDADE TUMORAL E DESENVOLVIMENTO DE FERRAMENTAS DE BIOINFORMÁTICA PARA ANÁLISE CITOGENÉTICA E MOLECULAR DE NEOPLASIAS SÓLIDAS. UNIVERSIDADE FEDERAL DO RIO GRANDE DO SUL INSTITUTO DE CIÊNCIAS BÁSICAS DA SAÚDE DEPARTAMENTO DE BIOQUÍMICA PROGRAMA DE PÓS-GRADUAÇÃO EM CB: BIOQUÍMICA. https://www.lume.ufrgs.br/bitstream/handle/10183/28744/000699142.pdf?sequence=1
-
Descrever a origem e o desenvolvimento da linhagem hematopoiética.
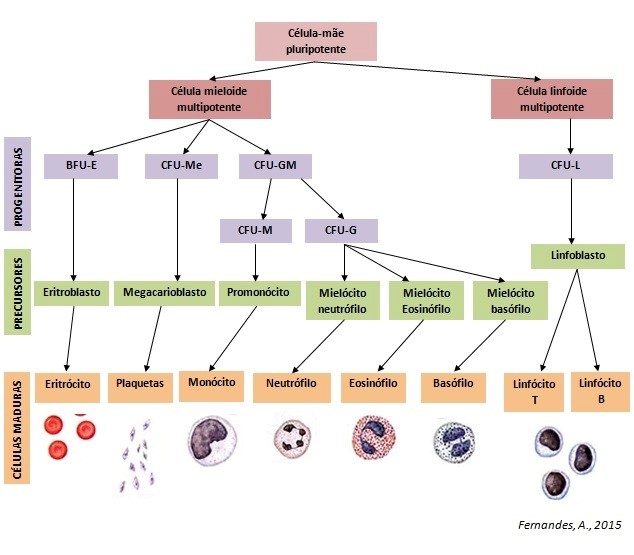
É o processo contínuo e regulado de produção de células do sangue que envolve renovação, proliferação, diferenciação e maturação celular. Ocorre a partir de um precursor celular comum e indiferenciado conhecido como célula hematopoiética pluripotente, célula-tronco ou stem-cell. As células-tronco, que no adulto encontram-se na medula óssea, são as responsáveis por formar todas as células e derivados celulares que circulam no sangue.
Sua origem se dá durante a 3ª semana de VIU no saco vitelínico. Durante o 3º mês, as células tronco hematopoiéticas (CTH) migram para o fígado, que se torna sítio principal de formação de células sanguíneas até o nascimento. A partir daí a medula óssea para a ser o sítio principal da hematopoese.
As CTH têm duas propriedades que a tornam capaz de manter a homeostase hematológica do organismo, ou seja, produzir o que for necessário e na medida certa:
- Pluripotência: que é a capacidade de uma única célula tronco dar origem a todas as demais.
- Autorrenovação: quando uma célula tronco se divide, ao menos uma deve migrar para os nichos especializados na medula, aonde receberão nutrientes e fatores necessários para tal. As demais células continuarão a ser estimuladas por fatores de transcrição e crescimento, sendo direcionadas, maturadas e morrendo ao final.
A hematopoiese é função do tecido hematopoiético, que aporta a celularidade e o microambiente tissular necessários para gerar os diferentes constituintes do sangue. No adulto, o tecido hematopoiético forma parte da medula óssea (medula vermelha) e é o local onde ocorre a hematopoiese normal.
O local onde ocorre a hematopoiese varia durante a ontogênese. Assim, observamos três fases sequenciais de locais hematopoiéticos:
- Fase mesoblástica: Fase inicial, no pedúnculo do tronco e saco vitelino. Ambas estruturas tem poucos mm. de longitude, ocorre na 2ª semana embrionária.
- Fase hepática: Na 6ª semana de vida embrionária.
- Fase mieloide: O baço e a medula óssea fetal.
3. Descrever a fisiopatologia das leucemias.
Neoplasias malignas do sistema hematopoiético caracterizadas pela proliferação clonal da célula-tronco, que progressivamente substitui a medula óssea normal e atinge o sangue periférico. Uma vez na circulação, as células neoplásicas podem infiltrar praticamente todas as vísceras, com preferência pelo fígado e baço.
Em todos os casos de leucemia, a medula óssea está comprometida, sendo variável a expressão da neoplasia no sistema periférico. Caracteriza-se pela proliferação predominante de células da série granulocítica, acompanhada ou não de proliferação de elementos da série megacariocítica.
- Fatores predisponentes para as Leucemias
- Translocações cromossômicas e outras Mutações adquiridas.
Os genes mutados ou que sofreram formas de alteração frequentemente apresentam papeis cruciais no desenvolvimento, crescimento ou sobrevivência da célula maligna.
As oncoproteínas criadas por aberrações genômicas muitas vezes bloqueiam a maturação normal.
Os proto-oncogenes são frequentemente ativados em células linfoides por erros que ocorrem durante a diversificação e o rearranjo gênico do receptor de antígenos.
- Fatores genéticos hereditários
Indivíduos com doenças genéticas que promovam instabilidade gênica, tais como a síndrome de Bloom, Anemia de Fanconi e a Atalaxia apresentam risco aumentado para o desenvolvimento de Leucemia Aguda.
- Vírus
EBV, HTLV1 e HHV-8 são linfonotrópicos e conhecidos cmo causadores de determinados linfomas.
- Estimulação Imune Crônica
Vários agentes ambientais que causam essa estimulação localizada predispõem à neoplasia linfoide, que quase sempre surge dentro do tecido inflamado. Ex: H. pylori e linfomas gástricos, sensibilidade ao glúten e enteropatias.
- Fatores Iatrogênicos
Radioterapia e outras formas de quimioterapia usadas no tratamento do câncer aumentam o risco de leucemias.
- Tabagismo
A incidência de LMA é até 2x maior em fuantes provavelmente pela exposição aos agentes carcinógenos, tais como o tabaco.
A doença começa assim que uma determinada célula progenitora (ao sofrer mutações genéticas) se torna incapaz de prosseguir na diferenciação hematopoiética. Esta célula não vai além da forma “jovem” (blasto) e começa a se proliferar descontroladamente, ocupando a medula óssea e impedindo o crescimento e a diferenciação das células normais. Devemos relembrar os passos da hematopoese, a fim de compreender melhor a fisiopatologia da leucemia aguda.
A célula tronco (célula totipotente) inicialmente se diferencia em dois tipos celulares (progenitores multilinhagem ou “CFU” – Colony Forming Units): um comprometido com a linhagem linfoide (formação dos linfócitos), e outro comprometido com a linhagem mieloide (formação de granulócitos, monócitos, hemácias e plaquetas).
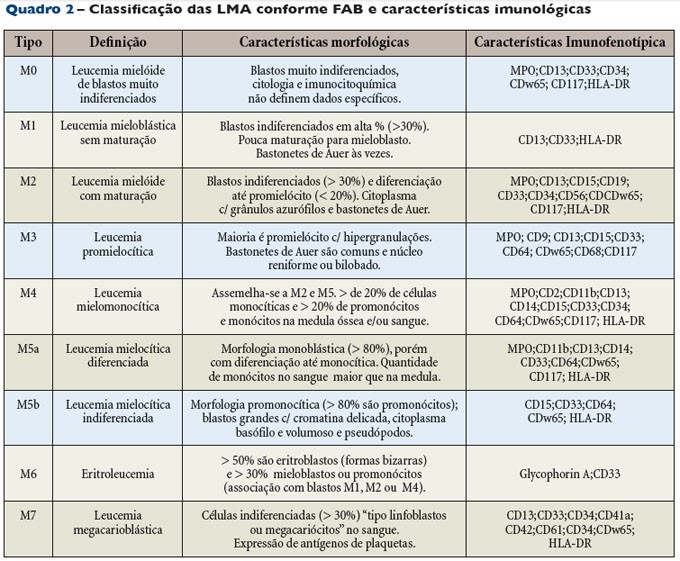
Os progenitores multilinhagem se diferenciam em progenitores de linhagem única, os quais se destinam a produzir um tipo celular especifico. Qualquer uma dessas células pode sofrer uma transformação neoplásica, inviabilizando o processo normal de maturação e resultando em proliferação e acumulo de um “clone” (população de células idênticas). Na LMA, o clone pode ter origem: na célula tronco, CFU-mieloide, CFU-GM ou CFU-E/mega, mieloblasto/pró-mielocito, monoblasto, eritroblasto ou megacarioblasto, entre outras células intermediarias. Repare que não citamos precursores “linfoides”, pois a leucemia aguda derivada destes é designada LLA (leucemia linfoide aguda). De forma bem simples podemos entender que cada subtipo morfológico de LMA corresponde ao tipo de célula que sofreu transformação. Assim, a morfologia do blasto irá caracterizar 8 subtipos de LMA, segundo a classificação da FAB.
Na LLA, como vimos, o clone neoplásico deriva de um progenitor linfoide, uma celula pré-T ou pré-B “precoce”, uma celula pré-T ou pré-B ou mesmo um linfócito B que assume características de blasto. Todas essas células são consideradas linfoblastos. Em 80% das LLA, a origem da neoplasia é na linhagem B. No restante (20%), a fonte é a linhagem de células T. Neste último caso, a leucemia pode cursar com proliferação de linfoblastos do timo, levando a uma entidade análoga ao linfoma linfoblastico, um tipo de linfoma não-Hodgkin de alto grau de malignidade, típico de crianças. Pela classificação da FAB, são 3 os subtipos morfológicos de LLA. Como veremos adiante, a identificação precisa do subtipo de LMA ou LLA é fundamental, pois tem implicações no quadro clinico, prognostico e tratamento.
Os blastos leucêmicos primeiramente infiltram a medula óssea, ocupando mais de 20% (pela OMS) ou mais de 30% (pela FAB) do total de células nucleadas, podendo chegar a 80-100% de ocupação. A primeira consequência, portanto, é a supressão da hematopoese normal. Essa expansão do clone neoplásico ocupa o espaço necessário a produção de células hematológicas normais, culminando em pancitopenia (anemia, leucopenia e plaquetopenia), o grande marco do início da doença. Os blastos anormais também secretam fatores inibitórios e indutores de fibrose, tornando a disfunção medular ainda mais grave do que o esperado somente pela ocupação do espaço. Esses blastos podem ser lançados na corrente sanguínea, justificando o termo leucemia (células brancas no sangue), com frequência atingindo um número suficientemente grande a ponto de determinar leucocitose. E como tais células não são capazes de amadurecer, elas não exercem qualquer função fisiológica. As defesas do organismo continuam dependendo dos poucos neutrófilos e monócitos existentes. Uma vez na corrente sanguínea, os blastos também podem infiltrar órgãos, com preferência para linfonodos, baço, fígado, gengiva, orbita, sistema nervoso central, meninges, testículos, pele, etc. O paciente vai ao óbito pela infiltração tecidual maciça (que leva a falência orgânica), e/ou pela pancitopenia grave e suas consequências (anemia, infecção, hemorragia).
- Tipagem e imunofenotipagem
Os blastos da LMA são um pouco maiores que os da LLA e geralmente apresentam grânulos azurófilos em seu citoplasma, o que define a linhagem granulocítica. A presença no citoplasma de filamentos eosinofílicos – os bastonetes de Auer (inclusões eosinofilicas em forma de agulha) – é patognomonica de LMA, embora presente apenas nos subtipos M1, M2, M3 e M4. Analisando apenas os aspectos morfológicos, um hematologista experiente consegue diferenciar a LMA da LLA em até 70% dos casos. A citoquímica ajuda nessa diferenciação. A coloração positiva para mieloperoxidase ou Sudan Black B indica LMA mieloblastica, e para esterase não especificas indica LMA monoblastica. A coloração positiva para PAS (ácido periódico de Schiff) sugere LLA derivada de células B. Já a coloração positiva para fosfatase acida indica LLA derivada de células T. Utilizando apenas a citoquímica, cerca de 15-20% dos blastos ainda continuam sem definição da origem.
Para definir com precisão o subtipo de célula leucêmica criou-se a imunofenotipagem, método considerado padrão-ouro para classificar as leucemias. O desenvolvimento dessa técnica foi uma das maiores revoluções no diagnóstico das neoplasias hematológicas. Consiste na pesquisa de marcadores na membrana ou citoplasma do blasto, através da administração de anticorpos específicos ligados a substancias fluorescentes. A positividade para determinado marcador é vista pela presença de atividade fluorescente na membrana ou citoplasma da célula neoplásica, diretamente ao microscópio, ou então – de preferência – por um processo automatizado chamado citometria de fluxo. Esses marcadores sao nomeados pela sigla CD (cluster designation). Cada subtipo de leucemia apresenta uma combinação própria de marcadores que o caracteriza. Por exemplo: os marcadores de membrana CD13, CD14 e CD33 definem a origem mieloide do blasto. O CD34 é um marcador de célula-tronco e confere pior prognostico. Ja o marcador TdT citoplasmático é detectado nos linfoblastos. Os marcadores de membrana CD10, CD19 e CD20 caracterizam o blasto linfoide. A presença do fator de Von Willebrand e Glicoproteina IIb/IIIa na superfície do blasto caracteriza a LMA-M7 (megacariocítica).
Hoje se sabe que por trás da maioria das neoplasias está a expressão de um ou mais oncogenes, isto é, genes responsáveis pela reprodução celular desordenada ou bloqueio de apoptose. Normalmente, oncogenes se localizam em regiões inativas do genoma, sendo denominados proto-oncogenes. Diversas formas de mutação podem transformar os proto-oncogenes em oncogenes. Os antioncogenes (genes supressores de tumor), por outro lado, são responsáveis pela síntese de fatores que inibem a expressão dos oncogenes. Eles podem ser inativados, o que facilita a expressão dos oncogenes.
Mutações frequentes identificadas nas leucemias agudas se associam a desarranjos estruturais nos cromossomos, e recebem a denominação genética de “anomalias citogenéticas”. Os principais exemplos são as deleções (perda de fragmentos), inversões (um fragmento cromossomial se inverte), translocações (troca de fragmentos entre dois cromossomos), ganhos cromossomiais (trissomias, hiperploidia) e perdas cromossomiais completas. O resultado final de tais fenômenos é a ativação de oncogenes ou supressão de antioncogenes.
As leucemias agudas podem ser primárias, quando surgem em um paciente sem nenhuma doença hematológica e sem uso prévio de quimioterápicos leucemogênicos, ou secundarias, quando surgem em pacientes com (1) doenças hematológicas pré-leucemicas, tais como síndromes mielodisplasicas (importante causa de LMA em idosos), síndromes mieloproliferativas, ou (2) uso prévio de certos quimioterápicos, como os agentes alquilantes e os inibidores de topoisomerase II. As leucemias secundarias sao via de regra do tipo LMA.
Os fatores de risco para as leucemias agudas fazem parte da sua etiopatogenia:
- Radiação ionizante: geralmente doses altas em indivíduos jovens, como radioterapia para tumores (LLA).
- Exposição a benzeno e derivados do petróleo (LMA).
- Agentes alquilantes (LMA): a leucemia se apresenta 4-6 anos após a exposição a droga e é quase sempre precedida por uma síndrome mieloplasica.
- Inibidores da topoisomerase II (LMA): a leucemia se apresenta precocemente (1-2 anos após exposição a droga) e nao se associa a síndrome mieloplasica. Esses fármacos também sao chamados de epipodofilotoxinas.
- Distúrbios hereditários (LMA): aqueles que levam a instabilidade cromossomial
- Anomalias citogeneticas congênitas: sindorme de down, síndrome de Patau.(trissomia do 13) e síndrome de klinefelter (XXY). A síndrome de down aumenta em 10-18 vezes a chance de leucemia aguda, concentrando seu pico etário na primeira infância. Até 3 anos de idade, predomina a LMA (subtipo M7), daí em diante, a LLA.
As leucemias agudas foram inicialmente classificadas pela FAB (French0American-British cooperative group), num esquema baseado em características morfológicas e citoquímicas dos blastos. Sua acurácia foi posteriormente reforçada com o advento da imunofenotipagem (citometria de pulso). Atualmente, entretanto, as decisões terapêuticas nas leucemias são bastantes dependentes de características nao contempladas pela classificação da FAB, como as anomalias citogenéticas e moleculares (que representam sindormes leucêmicas especificas), a relação com síndrome mielodisplásica prévia, uso de agentes alquilantes e inibidores da topoisomerase II.
Por isso, a OMS reclassificou (em 2008) as leucemias agudas sob uma ótica distinta, definindo grupos de prognostico e tratamento diferenciados. Cita também leucemias mais raras que não participam da classificação da FAB. Em suma, esta é a classificação mais utilizada nos dias atuais.
4. Explicar os sinais e sintomas, correlacionando-os com a patogênese e evolução das leucemias (destacando sua eventual semelhança com outras doenças menos graves).
- Manifestações clínicas – Leucemias Agudas
Os sinais e sintomas da leucemia aguda são geralmente de início rápido, desenvolvendo-se num período de poucas semanas até o máximo de uns poucos meses, e resultam da redução da função normal da medula óssea e da invasão de órgãos normais por blastos leucêmicos. A anemia está presente no diagnóstico na maioria dos pacientes e causa cansaço, palidez e cefaleia, e, nos pacientes predispostos, angina ou insuficiência cardíaca. Geralmente, encontramos trombocitopenia, e aproximadamente um terço dos pacientes apresenta sangramento clinicamente evidente no diagnóstico, em geral sob a forma de petéquias, equimoses, sangramento gengival, epistaxe ou hemorragia. A maioria dos pacientes portadores de leucemia aguda se encontra significativamente granulocitopênica no diagnóstico. Em consequência, aproximadamente um terço dos pacientes portadores de LMA e um número ligeiramente menor de pacientes portadores de LLA apresentam, na avaliação inicial, infecções significativas ou que causem risco de morte, a maioria das quais de origem bacteriana.
Além da supressão da função medular, as células leucêmicas podem infiltrar órgãos normais. Em geral, a LLA costuma infiltrar esses órgãos com maior frequência do que a LMA. O aumento dos linfonodos, fígado e baço é comum no momento do diagnóstico. A dor óssea, considerada como resultado da infiltração leucêmica no periósteo ou como expansão da cavidade medular, é uma queixa comum, especialmente nas crianças com LLA. Por vezes, as células leucêmicas infiltram a pele, resultando num exantema elevado, não pruriginoso, designado como cútis leucêmica. As células leucêmicas podem infiltrar as leptomeninges e causar meningite leucêmica, que se manifesta tipicamente por cefaleia e náuseas. Com a progressão da doença, podem se desenvolver paralisias do sistema nervoso central (SNC) e convulsões. Apesar do fato de menos de 5% dos pacientes com LLA terem envolvimento do SNC no diagnóstico, este é um local frequente de recidiva.
- Manifestações clínicas – Leucemias Crônicas
Cerca de 40% a 50% dos pacientes diagnosticados com LMC são assintomáticos até que a doença seja encontrada em exames físicos de rotina ou de sangue. O grau de leucocitose correlaciona-se à carga tumoral, definida pelo tamanho esplênico.
Os sintomas de LMC, quando presentes, são devidos à anemia e à esplenomegalia; incluem fadiga, perda de peso, mal-estar, saciedade fácil e dor ou sensação de plenitude no quadrante superior esquerdo. Raramente ocorrem sangramentos (associados à baixa contagem de plaquetas e/ou disfunção de plaquetas) ou trombose (associada à trombocitose e/ou leucocitose acentuada). Outras apresentações raras incluem artrite gotosa (devido a níveis elevados de ácido úrico), priapismo (geralmente com leucocitose ou trombocitose acentuadas), hemorragia de retina, ulceração e sangramento gastrointestinais altos (em razão de altos níveis de histamina pela basofilia). Dores de cabeça, dores ósseas, artralgias, dor por infarto esplênico e febre são incomuns na fase crônica, porém vão ficando mais frequentes com o progresso da LMC. Os sintomas leucostáticos, tais como dispneia, sonolência, perda de coordenação ou confusão, causados pela aglutinação de células em vasos pulmonares ou cerebrais, são incomuns na fase crônica, apesar de a contagem de leucócitos exceder 50.000 células/μL, embora estes sintomas possam aparecem mais frequentemente nas fases aceleradas ou blásticas.
A esplenomegalia, o sinal físico mais consistente na LMC, ocorre em 50% a 60% dos casos. A hepatomegalia já é menos comum (10% a 20%) e, em geral, de pouca amplitude (1 a 3 cm abaixo da margem costal direita). A linfadenopatia é incomum, assim como a infiltração da pele ou de outros tecidos. Caso presente, esses achados sugerem uma LMC Ph-negativa ou as fases acelerada ou blástica de LMC.
Referência: Goldman Cecil Medicina, Capítulo 189: Leucemias agudas| Lee Goldman, MD and Andrew I. Schafer, MD,https://www.evolution.com.br/epubreader/goldman-cecil-medicina-24ed
Goldman Cecil Medicina, Capítulo 190: As leucemias crônicas| Lee Goldman, MD and Andrew I. Schafer, MD,https://www.evolution.com.br/epubreader/goldman-cecil-medicina-24ed.
5. Citar as diversas leucemias, suas formas de apresentação e sua epidemiologia, relacionando: tipo de leucemia, faixa etária e prognóstico.
- Epidemiologia Geral – Fonte: INCA
- Estimativas de novos casos: 10.070, sendo 5.540 homens e 4.530 mulheres (2016 – INCA).
- Número de mortes: 6.316, sendo 3.439 homens e 2.877 mulheres (2013- SIM).
O que caracteriza as leucemias “agudas” é a ocorrência de um acúmulo de progenitores de linhagem linfoide ou mieloide, células que recebem a denominação de blastos (blasto = célula jovem). Os blastos são incapazes de se diferenciar em células maduras, devido a um bloqueio de maturação, o grande marco fisiopatológico da doença. As leucemias crônicas, em contrapartida, são caracterizadas pelo acumulo de células maduras ou quase maduras. Estas podem ser derivadas de clones neoplásicos mais jovens, que seguiram o processo normal de maturação.
Faixa etária
- Adulto: A forma mais comum de leucemia aguda é a LMA (leucemia mieloide aguda).
- Na criança com menos 15 anos de idade, as leucemias quase sempre são agudas, sendo a mais frequente a LLA (leucemia linfoblástica aguda) – na realidade, a LLA é o tipo mais comum de câncer (em geral) da criança.
- Agora, considerando o grupo das leucemias como um todo (agudas e crônicas), o tipo mais comum de leucemia no mundo é a LLC (leucemia linfocítica crônica).
Leucemia Mieloide Aguda (LMA)
É um tumor de progenitores hematopoiéticos causados por mutações oncogênicas adquiridas que impedem a diferenciação, resultando acumulação de blastos mieloides na medula.
É a leucemia aguda mais comum, afetando uma faixa etária bastante ampla. Sua incidência começa a se elevar a partir dos 15 anos e tende a aumentar progressivamente com a idade. Assim, um adulto com leucemia aguda provavelmente tem LMA. Sabemos que a doença tem ligeira preferência pelo sexo masculino.
- Início abrupto
- Sinais e sintomas relacionados à falência da hematopoese normal: anemia refratária, neutropenia (infecções) e (hemorragia) trombocitopenia. Hepatoesplenomegalia não é muito comum.
- Febre, petéquias, equimoses cutâneas.
A evolução dos sintomas pode ser aguda ou subaguda (semanas), embora metade dos pacientes apresente queixas inespecíficas nos últimos 3 meses. A tríade sintomática da leucemia aguda é: astenia, hemorragia e febre, todos sintomas relativos a insuficiência hematopoiética medular. Essa tríade é a mesma da anemia aplasica, sendo este o diagnostico diferencial mais importante, principalmente quando não há leucocitose no hemograma.
A astenia, ou fadiga, é o sintoma inicial em metade dos casos. A astenia, na verdade, é o principal componente da síndrome anêmica. Estes pacientes normalmente desenvolvem uma anemia moderada a grave instalação rápida. Os outros comemorativos da síndrome anêmica também podem estar presentes: dispneia, cefaleia e tontura postural.
O sangramento reflete a plaquetopenia grave e, eventualmente, um distúrbio da coagulação na LMA. Quando o distúrbio é secundário apenas a plaquetopenia, manifesta-se com sangramento cutâneo (petéquias, equimoses) e mucoso (sangramento gengival, epistaxe, metrorragia, hemorragia digestiva). Em alguns casos, a diátese (pré-disposição para algumas doenças) hemorrágica é desproporcional ao grau de plaquetopenia, em razão da coexistência de disfunção das plaquetas circulantes.
A febre pode ser decorrente de dois mecanismos: neutropenia/ disfunção neutrofílica, que favorece infecções bacterianas sistêmicas – mecanismo mais comum, ou febre neoplásica consequente a rápida proliferação clonal.
Outros sinais e sintomas são decorrentes da infiltração leucêmica de órgãos e tecidos. Vejamos alguns exemplos. A hepatoesplenomegalia é uma manifestação frequente e pode diferenciar clinicamente a leucemia aguda de uma anemia aplasica (que não cursa hepatoesplenomegalia). A esplenomegalia das leucemias agudas não é tão proeminente quanto a da LMC.
A dor óssea, também mais comum na LLA, é um sintoma decorrente da expansão medular pela proliferação dos blastos ou da invasão do periósteo.
Quando a leucometria alcança valores exorbitantes (>50.000 ou >100.000/mm3) – uma condição conhecida como hiperleucocitose – a síndrome da leucostase pode se instalar. Os leucócitos aumentam a viscosidade sanguínea e podem se aderir ao endotélio das vênulas pulmonares e outros órgãos, como o cérebro. O paciente apresenta sintomas neurológicos (cefaleia, borramento visual, parestesia, etc), pulmonares (dispneia, taquipneia, insuficiência respiratória com hipoxemia grave) e geniturinários (priapismo (ereção dolorosa), insuficiência renal aguda). Casos mais graves devem ser imediatamente tratados com leucoaférese (retirada de leucócitos do sangue).
O hemograma de uma leucemia aguda é caracterizado pela presença de anemia + plaquetopenia, com leucometria variável. A anemia é um achado quase universal, sendo geralmente moderada a grave (Hb entre 5-9g/dL), normocitica, normocromica e sem reticulocitose. O grau de plaquetopenia varia, com cerca de 25% dos pacientes possuindo plaquetas abaixo de 20.000/mm3.
A leucocitose é um achado comum, embora alguns casos abram com leucopenia. A leucocitose é representada por blasto na periferia, geralmente associada a neutropenia.
Quase sempre os blastos são encontrados no esfregaço de sangue periférico, podendo ser contados no hemograma. Entretanto, numa minoria, os blastos estão ausentes na periferia, sendo encontrados apenas na medula óssea, uma condição denominada “leucemia aleucemica”.
O aumento dos níveis séricos de lisozima é característico dos subtipos M4 e M5 e pode ter consequencias clinicas, como a lesão tubular renal, levando a insuficiência renal aguda. A pseudo-hipercalemia e a pseudo-hipoglicemia podem ocorrer especialmente no laboratório (os blasto no tubo de ensaio liberam potássio e consomem glicose).
- Diagnóstico
- Hemograma: leucocitose (blastos), ↓eritrócitos, Hb, plaquetas e neutrófilos.
- Mielograma: mínimo 20% blastos na MO (bastão de Auer)
- Biópsia: idem mielograma
- Imunofenotipagem: HLA-DR, CD35 e CD13.
- Citogenética: alterações cromossômicas.
Diagnóstico deve ser sempre confirmado pelo mielograma (aspirado de medula óssea), obtido geralmente da crista ilíaca. É necessária a presença de mais de 20% de blastos (OMS) entre as células nucleadas do aspirado. Além de confirmar uma leucemia aguda, o exame deve tipar e subtipar a leucemia, definindo dados prognósticos. A biopsia de medula óssea também deve ser realizada para analise das alterações displasicas e do grau de mielofibrose associada.
- Transfusão (plaquetopenia e anemia)
- Terapia especifica de LMA (deve ser feita com quimioterapia de alto poder mielotóxico, para tentar destruir completamente o clone mieloblástico. O transplante halogênico de células hematopoiéticas é um importante item da terapia, sendo a única chance de cura em alguns pacientes)
50% sobrevivência (mau prognóstico)
Leucemia Linfoide Aguda (LLA)
É a leucemia mais comum na infância. Um pico de incidência entre 2-10 anos é registrado. É mais comum na raça branca e tem discreta predominância no sexo masculino. A LLA também pode ocorrer no adulto. Nesse caso, a doença apresenta pior prognostico, com uma taxa de cura de apenas 25 a 40%. Os adultos respondem bem a quimioterapia de indução com até 90% de remissão completa, a maioria experimenta recaída da doença.
São neoplasias compostas de células B (pré-B) ou T (pré-T) imaturas as quais são chamadas de linfoblastos.
Alterações cromossômicas: Hiperploidia (>50 cromossomos), hipoploidia (Muitas dessas alterações desregulam a expressão e a função dos fatores de transcrição necessários para o funcionamento normal de células B e T, interrompendo sua maturação.
LLA-B: mutação com ganho de função no gene NOTCH1 (desenvolvimento de células T)
LLA-T: mutação com perda de função nos genes: PAX-5, E2A e EBF (desenvolvimento de células B) ou uma t (12;21) nos genes TEL e AML1 (precursores hematopoiéticos na fase inicial do desenvolvimento).
- Início repentino e violento
- Depressão funcional da M.O: fadiga e anemia (↓hemácias), hemorragia (trombocitopenia), hepatoesplenomegalia.
- Infiltração neoplásica: dor óssea e subperiósteo, linfadenopatia.
- Alterações do SNC: cefaleia, vômito e paralisia dos nervos (expansão meníngea)
O quadro clínico da LLA é muito semelhante ao da LMA. Contudo, algumas diferenças devem ser destacadas: a dor óssea é muito frequente – 80% dos casos; adenomegalia cervical ou generalizada é mais frequente – 75% dos casos; podem ocorrer massas mediastinais no subtipo D células-T do timo; o acometimento do sistema nervoso central e dos testículos é mais comum; febre neoplásica é mais comum – 70% dos casos; a hiperplasia gengival não faz parte do quadro clinico.
- Hemograma: leucocitose (variável), plaquetopenia (< 100 mil), anemia (Hb < 10) g/dl), neutropenia e trombocitopenia (casos graves).
- Mielograma: blastos ( >25%)
- Biópsia: (> 25%)
- Citogenética: alterações cromossômicas
- Imunofenotipagem: fenótipos blásticos.
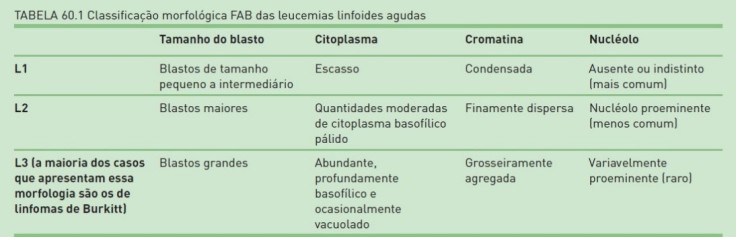
O critério diagnóstico para LLA é a presença de linfoblastos na medula óssea em proporção maior ou igual a 25% do total de células nucleadas. A diferenciação entre linfoblastos e blastos da LMA (mieloblastos) é feita por critérios morfológicos, citoquímicos e imunofenotipagem. Pelos critérios da FAB existem três subtipos de LLA.
- O subtipo L1 é o mais comumente encontrado na LLA infantil (80% dos casos), apresentando o melhor prognostico e resposta a terapêutica. O linfoblasto possui núcleo arredondado e citoplasma escasso.
- O subtipo L2 é a forma mais comum no adulto. O linfoblasto é maior que o L1, tem nucléolos, mais citoplasma e núcleos irregulares.
- O subtipo L3 é o menos comum, e representa a forma leucêmica do linfoma de Burkitt, caracterizado por linfoblastos de tamanho intermediário, com citoplasma proeminente, basofilico e cheio de vacúolos
- Tratamento
- Transplante de células hematopoiéticas.
- Medicamentos (ATRA – t(15;17))
- Quimioterapia
- Correção de Hemograma
- Prognóstico
- Positivo: 2-10 anos
- Negativo: rearranjo MML ou t(9;22) Ph
Leucemia Mieloide Crônica (LMC)
É uma síndrome mieloproliferativa crônica, juntamente com a policitemia vera, metaplasia mieloide agnogênica e a trombocitemia essencial. As síndromes mieloproliferativas formam um grupo de neoplasias hematológicas que se originam da célula-tronco ou de um progenitor próximo a esta em sua maturação. Porém, ao contrário das leucemias agudas, esse clone segue curso normal de maturação até as células finais (granulócitos, hemácias e plaquetas) – NÃO HÁ BLOQUEIO DE MATURACAO. O pico de incidência da LMC é na fase adulta, entre 45-55 anos, porem pode ocorrer em crianças. Existe um discreto predomínio no sexo masculino.
O clone neoplásico da LMC provavelmente é uma célula-tronco. Por razões desconhecidas essas células adquirem uma anomalia citogenética denominada cromossomo Philadelfia que, na verdade, é uma translocação entre os braços longos do cromossomo 9 e 22. A posição desses genes forma um oncogene hibrido, responsável pela síntese de proteína P210 incriminada no aumento da divisão celular e no bloqueio da apoptose. Cerca de 95% dos pacientes com LMC apresenta cromossomo Philadelfia detectável na análise do cariótipo das células do aspirado de medula óssea. O clone neoplásico é capaz de se diferenciar em células maduras, diferente do encontrado nas leucemias agudas. A diferenciação ocorre preferencialmente na medula óssea e no sangue periférico de neutrófilos, bastões, metamielocitos, mielocitos e, eventualmente, raros mieloblastos. Os eosinófilos e basófilos encontram-se elevados. Os monócitos e plaquetas podem se elevar, porém, a hematimetria tende a se reduzir (anemia) por conta da ocupação medular neoplásica, inibindo a eritropoiese.
- Manifestações clínicas e laboratoriais
Caracterizado por hiperplasia medular e capacidade de maturação das células mieloides.
Sintomais: anemia branda/moderada, hipermetabolismo celular resultando em fadiga, perda de peso, anorexia, desconforto abdominal (esplenomegalia).
- Fase Acelerada (3-18 meses)
Perda progressiva de diferenciação celular (A LMC está associada a mutações que interferem no Ikaros, fator de transcrição que regula a diferenciação dos progenitores hematopoiéticos.)
Sintomas: Acentuada anemia, trombocitopenia, as vezes com mais basófilos.
Quadro semelhante à leucemia aguda. Presença de mais de 20% de blastos no sangue periférico ou medula.
Muitos pacientes com LMC são descobertos em uma fase assintomática da doença, através do exame físico mostrando esplenomegalia e/ou hemograma revelando leucocitose neutrofílica acentuada, com desvio para esquerda até mielocito ou mieloblasto. O marco da LMC é justamente essa associação (leucocitose neutrofílica acentuada com desvio a esquerda + esplenomegalia).
Os sintomas mais comuns de apresentação da doença são decorrentes do estado hipercatabólico, da esplenomegalia, da anemia e/ou da disfunção plaquetária, tais como: febre, perda ponderal, astenia, sudorese noturna, desconforto abdominal no hipocôndrio esquerdo, saciedade precoce, palpitação, dispneia, equimoses. As infecções na LMC não são frequentes, nem caracterizam a doença. O clone neoplásico é capaz de se diferenciar até o neutrófilo maduro. Este neutrófilo possui uma função normal ou levemente diminuída.
Quadro laboratorial da doença caracteriza-se pela acentuada leucocitose neutrofílica, invariavelmente presente. O diferencial revela intenso desvio para esquerda, havendo muitas formas jovens granulocíticas na periferia: bastões, metamielocitos, mielocitos e até mieloblastos. A contagem absoluta de eosinófilos e basófilos está via de regra alta. A LMC É UMA DAS UNICAS CAUSAS DE BASOFILIA PROEMINENTE E PERSISTENTE.
A regra portanto, na LMC, é: anemia + hiperleucocitose + trombocitose
- Hemograma: Leucocitose/granulócitos, trombocitose, ↓hemácias
- Mielograma: ↑blastos (fase blástica), ↓blastos (fase crônica)
- Citogenética: Ph t(9;22).
- Anamnese e exame físico;
- Plaquetometria;
- Morfologia de sangue periférico;
- Fosfatase alcalina dos neutrófilos no sangue periférico;
- Citoquímica e Imunofenotipagem (apenas na FB);
- Citogenética da medula óssea;
- PCR qualitativo: pesquisa do marcador molecular
- BCR/ABL no sangue periférico ou na medula óssea;
- Biópsia de medula óssea incluindo determinação de fibrose medular.
Deve ser suspeitado sempre que houver leucocitose acentuada (> 25.000-50.000/mm3) e esplenomegalia. O diagnóstico diferencial deve ser feito sempre com as reações leucemoides, com as outras síndromes mieloproliferativas.
Nos casos suspeitos, um aspirado (mielograma) e uma biópsia de medula óssea devem ser realizados. A CONFIRMACAO DIAGNOSTICA É FEITA PELO ACHADO DO CROMOSSOMO PHILADELFIA NA AVALIACAO CITOGENETICA DAS CELULAS DO ASPIRADO.
- Correção de plaquetopenia e anemia: hemocomponentes filtrados.
- Quimioterapia e corticoides
- Transplante de MO: em casos de +Ph ou doença recidiva.
- Inibidores de tyrosine kinase (faz com que a célula siga seu caminho normal para a apoptose) e transplante medular.
Leucemia Linfoide Crônica (LLC)
São neoplasias clonais de linfócitos maduros não competentes, resistentes à morte que se acumulam em órgãos linfoides. Ao contrário da LMC, não existe uma única anomalia cromossomial típica da LLC, apesar de existirem algumas alterações que modificam o prognostico da doença. A evolução da doença é o acúmulo de linfócitos B neoplásicos na medula óssea, passando em seguida para o sangue periférico e atingindo os linfonodos, baço e fígado. O paciente vai se tornando debilitado e extremamente propenso a morrer de infecções bacterianas.
A LLC é a segunda leucemia mais comum e acomete caracteristicamente a população idosa, sendo a mais comum leucemia nessa faixa etária, com preferência 2:1 no sexo masculino. A LLC praticamente não é vista em pessoas com menos de 30 anos de idade e não acomete crianças. Trata-se de uma neoplasia hematológica cujo clone neoplásico é um linfócito B maduro, porém bloqueado numa fase de diferenciação, que impede a sua transformação em plasmócito, a célula produtora de anticorpos.
- Manifestações clínicas e laboratoriais
- Os pacientes são frequentemente assintomáticos ao diagnóstico mas quando aparecem são inespecíficos.
- Encontramos: hepatoesplenomegalia, sudorese, anorexia, fadiga, linfadenopatia e infecções recorrentes. Portanto sinais de falência medular.
Muitos pacientes são diagnosticados na fase assintomática da doença, pelo encontro de uma linfocitose expressiva no hemograma. Linfocitose é definida por mais de 3.000 linfócitos por mm3 no hemograma. Depois da linfocitose, a adenomegalia cervical (aumento dos linfonodos do pescoço) é o achado mais comum, presente em 2/3 dos pacientes na apresentação da doença. O marco da LLC é a associação: linfocitose acentuada + adenomegalia.
- Hemograma: leucocitose, trombocitopenia e anemia (↓hemácias)
- Mielograma: linfoblastos
- Biópsia: se necessário
- Imunofenotipagem: CD5, CD23, CD38, CD19, CD20, ZAP.
- Citogenética: Deleções 13q, 11q.
Diagnostico confirmado por um dos critérios:
- Linfocitose persistente > 10.000/mm3 + aspirado de medula óssea com>30% de linfócitos.
- Linfocitose persistente > 5.000/mm3 + aspirado de medula óssea com >30% de linfócitos + imunofenotipagem revelando marcadores de linfócito B maduro em conjunto com marcador CD5.
Transplante medular
A sobrevida geral é de 4-6 anos, logo mau prognóstico.
Referência:
- UNICAMP/FOP. Áreas de semiologia e patologia.
- http://w2.fop.unicamp.br/ddo/patologia/downloads/db301_un5_Aula49Linfo-Leucemia2.pdf
- INCA http://www1.inca.gov.br/rbc/n_49/v01/pdf/condutas.pdf
6. Descrever os exames utilizados para o diagnóstico e estadiamento de leucemias.
Leucemia Mieloide Crônica (LMC)
- Hemograma completo (exame de sangue): a doença provoca aumento significativo dos glóbulos brancos, e também pode atingir os glóbulos vermelhos e as plaquetas, e essas alterações serão constatadas neste exame. O aumento do baço, comum a alguns pacientes, também pode ser verificado em exames clínicos e de imagem.
- Mielograma ocorre quando uma amostra de sangue da medula óssea é retirada por meio de uma agulha. É um exame de grande importância para o diagnóstico (análise das células) e para a avaliação da resposta ao tratamento, indicando se, morfologicamente, essas células leucêmicas foram erradicadas da medula óssea (remissão completa medular). Esse exame é feito sob anestesia local e consiste na aspiração da medula óssea seguida da confecção de esfregaços em lâminas de vidro, para exame ao microscópio. Os locais preferidos para a aspiração são a parte posterior do osso ilíaco (bacia) e o esterno (parte superior do peito). Durante o tratamento são feitos vários mielogramas.
- Os exames de citogenética, como o cariótipo, são fundamentais, pois avaliam especificamente os cromossomos (a busca é pelo cromossomo Philadelphia para fechar o diagnóstico).
- Já o FISH (Hibridização Fluorescente in situ) é outro tipo de exame que avalia os cromossomos, e detecta as alterações pequenas não visualizadas.
- Há ainda o teste molecular de reação em cadeia da polimerase (PCR), utilizado para o diagnóstico e acompanhamento da doença. Ele mede, no sangue ou medula óssea, a quantidade do gene de fusão causador deste tipo de câncer, o BCR-ABL.
**De todos estes exames, o único que não está disponível no Sistema Único de Saúde é o FISH. Porém, ele pode ser feito com o plano de saúde.
O estadiamento da maioria dos tipos de cânceres atribui estágios numerados, de I a IV, para descrever sua extensão, com base no tamanho do tumor e na probabilidade de disseminação.
A leucemia mieloide crônica é uma doença da medula óssea e não segue um sistema de estadiamento como a maioria dos cânceres. O prognóstico de um paciente com leucemia mieloide crônica depende de outras informações, como estágio da doença, idade, exames de sangue e valores sanguíneos e comprometimento do baço.
Fases da Leucemia Mieloide Crônica
A leucemia mieloide crônica é dividida em 3 grupos, baseada principalmente no número de glóbulos brancos imaturos (mieloblastos):
Os pacientes nesta fase têm menos do que 10% de blastos nas amostras de sangue ou medula óssea. Estes pacientes geralmente apresentam sintomas leves (se existirem) e geralmente respondem ao tratamento padrão. A maioria dos pacientes é diagnosticada na fase crônica.
Os pacientes são considerados em fase acelerada, se qualquer um dos seguintes for verdadeiro:
- Amostras de sangue ou da medula óssea com mais de 10% e menos do que 20% de blastos.
- Alta taxa de basófilos no sangue (pelo menos, 20% das células brancas do sangue).
- Alta taxa de glóbulos brancos, que não diminui com o tratamento.
- Contagem de plaquetas muito alta ou muito baixa, não causadas pelo tratamento.
- Novas alterações cromossômicas nas células leucêmicas.
Os pacientes com leucemia mieloide crônica em fase acelerada podem apresentar sintomas como febre, falta de apetite e perda de peso.
A medula óssea e as amostras de sangue de um paciente nesta fase têm mais de 20% de blastos. As células blásticas frequentemente se espalham para tecidos e órgãos além da medula óssea. Esses pacientes geralmente apresentam sintomas, como febre, falta de apetite e perda de peso.
Fatores Prognósticos
As diferenças entre os pacientes e que afetam a resposta ao tratamento são denominados fatores prognósticos. Alguns fatores são vinculados com um menor tempo de sobrevida e são denominados fatores prognósticos adversos.
- Fase acelerada ou fase blástica.
- Aumento do baço.
- Áreas de dano ósseo devido ao avanço da leucemia.
- Aumento do número de basófilos e eosinófilos em amostras de sangue.
- Contagem de plaquetas muito alta ou muito baixa.
- Idade (60 anos ou mais).
- Múltiplas alterações cromossômicas nas células leucêmicas.
Muitos destes fatores são levados em conta no sistema de Sokal, que desenvolve uma pontuação utilizada para o prognóstico do paciente. Este sistema considera a idade, a porcentagem de blastos no sangue, o tamanho do baço e a quantidade de plaquetas. Estes fatores são utilizados para classificar os pacientes em grupos de risco (baixo, intermediário ou alto). Outro sistema, denominado sistema Euro inclui além dos fatores mencionados acima, o número de basófilos e eosinófilos. Ter grandes quantidades dessas células indica um pior prognóstico.
Os sistemas Sokal e Euro foram úteis no passado, antes do desenvolvimento de medicamentos mais eficazes para a leucemia mieloide crônica. No momento, não está claro como eles podem ser úteis na determinação do prognóstico de um paciente.
As terapias alvo como o imatinib mudaram drasticamente o tratamento da leucemia mieloide crônica nos últimos anos. Esses sistemas não foram testados em pacientes tratados com estes medicamentos.
Leucemia Linfoide Crônica (LLC)
- Exames
- Hemograma (exame de sangue).
- Mielograma(com uma agulha, é coletada uma pequena quantidade de sangue da medula),
- Biópsia da medula(quando é retirado, com uma agulha, um pequeno fragmento da região da medula óssea) que irão mostrar as características dos glóbulos brancos.
- Os testes deimunofenotipagem e de citogenética (cariótipo), feitos com uma pequena amostra de sangue, irão analisar as células de maneira bem específica e serão os responsáveis pelo diagnóstico do tipo da leucemia – no caso, a leucemia linfoide crônica.
- O FISH(hibridização por fluorescência in situ), exame bastante sensível, que por meio de uma pequena amostra de sangue, pode detectar uma célula anormal em meio a 700 células normais.
- Acitometria de fluxo pode ser outra opção pedida pelo médico, para revelar a presença da leucemia linfoide crônica, pois este aparelho consegue medir de maneira individual milhares de células.
- Em raros casos, o paciente pode apresentar aumento nos gânglios linfáticos (carocinhos, que aparecem na região da virilha, pescoço e axilas). Se isso ocorrer, o médico deverá pedir uma biópsia do gânglio, para uma melhor avaliação.
O estadiamento da maioria dos tipos de cânceres atribui estágios numerados para descrever sua extensão, com base no tamanho do tumor e na probabilidade de disseminação.
A leucemia linfocítica crônica por outro lado, normalmente não forma massas tumorais, mas geralmente afeta toda a medula óssea e, em muitos casos, pode se espalhar para outros órgãos, como o fígado, baço e gânglios linfáticos. Por conseguinte, a perspectiva para o paciente com leucemia linfoide crônica depende de outras informações, como subtipo da leucemia linfoide crônica, idade, e resultados de exames de laboratório.
Um sistema de estadiamento é a maneira padronizada para que todos os membros de uma equipe multidisciplinar entendam de imediato a extensão da doença. Existem 2 sistemas utilizados na LLC: o sistema Rai e o sistema Binet.
Sistema de Estadiamento Rai
O sistema RAI divide a leucemia linfoide crônica em 5 estágios:
- Estágio Rai 0 – Linfocitose (contagem de linfócitos no sangue muito elevada).
- Estágio Rai I – Linfocitose mais linfonodos aumentados. O baço e o fígado não estão aumentados e glóbulos vermelhos e plaquetas normais.
- Estágio Rai II – Linfocitose e aumento do baço (e, possivelmente, aumento do fígado), com ou sem aumento dos gânglios linfáticos. Glóbulos vermelhos e plaquetas normais.
- Estágio Rai III – Linfocitose mais anemia, com ou sem aumento dos gânglios linfáticos, baço ou fígado. Plaquetas normais.
- Estágio Rai IV – Linfocitose mais trombocitopenia, com ou sem anemia, aumento dos gânglios linfáticos, baço ou fígado.
Para fins práticos, os médicos separam os estágios Rai em 3 grupos:
- Estágio 0 – Risco baixo.
- Estágio I e II – Risco intermediário.
- Fases III e IV – Risco alto.
Sistema de Estadiamento Binet
No sistema de estadiamento Binet, a leucemia linfoide crônica é classificada pelo número de grupos de tecido linfoide afetados (linfonodos cervicais, linfonodos inguinais, linfonodos axilares, baço e fígado) e pelo fato do paciente apresentar anemia ou trombocitopenia:
- Estágio Binet A – Menos do que 3 áreas de tecido linfoide aumentadas, sem anemia ou trombocitopenia.
- Estágio Binet B – 3 ou mais áreas de tecido linfoide aumentadas, sem anemia ou trombocitopenia.
- Estágio Binet C – Anemia ou trombocitopenia presente.
Recentemente, os médicos entenderam que outros fatores também podem ajudar a prever o prognóstico de um paciente. Os fatores descritos a seguir não fazem parte de nenhum sistema de estadiamento formal atualmente, mas eles também podem fornecer informações úteis.
Existem outros fatores que ajudam a definir o prognóstico:
Fatores Prognóstico Adversos – Padrão difuso de envolvimento da medula óssea; idade avançada; sexo masculino; exclusões de partes de cromossomos 17 ou 11; altos níveis sanguíneos de substâncias, como beta-2-microglobulina; tempo de duplicação dos linfócitos menor que 12 meses; aumento da proporção dos linfócitos grandes ou atípicos no sangue; elevada percentagem de células que contêm ZAP-70 ou CD38; e, células com gene inalterado para imunoglobulina de cadeia pesada na região variável (IGHV).
Fatores Prognóstico Favoráveis – Padrão não difuso (nodular intersticial) do envolvimento da medula óssea; supressão de parte do cromossomo 13 (sem outras anormalidades cromossômicas); baixa proporção de células que contêm ZAP-70 ou CD38; e, e, células com gene inalterado para imunoglobulina de cadeia pesada na região variável (IGHV).
Os fatores prognóstico com base em testes de laboratório mais recentes, como a presença ou ausência de ZAP-70 e CD38, provavelmente se tornarão mais importantes ao longo do tempo, e podem, eventualmente, serem mais assertivos, particularmente, para pacientes em estágios iniciais da leucemia linfoide crônica.
Leucemia Mieloide Aguda (LMA)
- Hemograma completo (exame de sangue).
- Mielograma.
- Ocariótipo, exame responsável pelo estudo das alterações cromossômicas, deve ser feito para uma melhor classificação da LMA.
- Já o FISH (Hibridização Fluorescente in Situ).
- Abreviatura de reação em cadeia da polimerase, oPCR quantitativo também tem sido uma importante ferramenta para o diagnóstico da doença.
- Os exames de citogenética,imunofenotipagem e biologia molecular, feitos com uma amostra de sangue do paciente, também são importantes, pois, de forma geral, avaliam especificamente os cromossomos e o grau de resistência da doença, facilitando ao especialista a escolha pelo tratamento ideal.
- Outro fator comum é o aumento do tamanho do baço e do fígado, notado por exames de imagem, comoradiografia de tórax, tomografia computadorizada, ressonância magnética e ultrassom.
- O uso deanticorpos monoclonais (proteínas usadas pelo sistema imunológico para identificar e neutralizar corpos estranhos como bactérias, vírus ou células tumorais. Eles reconhecem um alvo específico, o antígeno, presente nas células estranhas ao organismo) reforça o diagnóstico de alguns casos, assim como permite a identificação da leucemia bifenotípica (quando a leucemia contém as características linfoides e mieloides ao mesmo tempo).
A leucemia mieloide aguda por outro lado, normalmente não forma massas tumorais, mas geralmente afeta toda a medula óssea e, em muitos casos, pode se espalhar para outros órgãos, como o fígado, baço e gânglios linfáticos. Por conseguinte, a perspectiva para o paciente com LMA depende de outras informações, como: subtipo da leucemia, idade e resultados de exames de laboratório.
É importante saber o subtipo da leucemia mieloide aguda, uma vez que influencia na escolha do tipo de tratamento e no prognóstico do paciente. Por exemplo, o subtipo leucemia promielocítica aguda é tratada com medicamentos diferentes dos utilizados para outros subtipos da leucemia mieloide aguda.
Os principais sistemas utilizados para estadiar a leucemia mieloide aguda são o sistema de estadiamento britânico-americano-francês e o mais recente o sistema de estadiamento da Organização Mundial de Saúde (OMS).
Sistema de Estadiamento Britânico-Americano-Francês
Na década de 1970, um grupo de especialistas franceses, americanos e britânicos dividiram a leucemia mieloide aguda em subtipos, M0 a M7, com base no tipo de célula em que a leucemia se desenvolve e o grau de maturidade das células.
|
FAB
|
Nome
|
|
M0
|
Leucemia mieloblástica aguda indiferenciada |
|
M1
|
Leucemia mieloblástica aguda com maturação mínima |
|
M2
|
Leucemia mieloblástica aguda com maturação |
|
M3
|
Leucemia promielocítica aguda |
| M4 |
Leucemia mielomonocítica aguda
|
| M4 eos |
Leucemia mielomonocítica aguda com eosinofilia
|
| M5 |
Leucemia monocítica aguda
|
| M6 |
Leucemia eritroide aguda
|
| M7 |
Leucemia megacarioblástica aguda
|
Os subtipos M0 a M5 se iniciam em formas imaturas dos glóbulos brancos. O subtipo M6 começa em formas muito imaturas dos glóbulos vermelhos, enquanto o M7 começa em formas imaturas das células produtoras das plaquetas.
O sistema de classificação FAB é útil e ainda é comumente utilizado para agrupar a leucemia mieloide aguda em subtipos. Mas, ele não leva em conta muitos fatores conhecidos, atualmente, por influenciar no prognóstico do paciente.
Sistema de Estadiamento da Organização Mundial da Saúde
A Organização Mundial da Saúde (OMS) desenvolveu um sistema mais novo que inclui alguns desses fatores para obter um melhor estadiamento da leucemia mieloide aguda.
O sistema de estadiamento da OMS divide a leucemia mieloide aguda em vários grupos:
- Leucemia Mieloide Aguda com Anormalidades Genéticas
- Leucemia mieloide aguda com uma translocação entre os cromossomos 8 e 21.
- Leucemia mieloide aguda com uma translocação ou inversão no cromossomo 16.
- Leucemia mieloide aguda com uma translocação entre os cromossomos 9 e 11.
- Leucemia profolítica aguda (M3) com uma translocação entre os cromossomos 15 e 17.
- Leucemia mieloide aguda com uma translocação entre os cromossomos 6 e 9.
- Leucemia mieloide aguda com uma translocação ou inversão no cromossomo 3.
- Leucemia mieloide aguda com uma translocação entre os cromossomos 1 e 22.
- Leucemia Mieloide Aguda com alterações relacionadas à Mielodisplasia
- Leucemia Mieloide Aguda relacionada a Quimioterapia ou Radioterapia Prévia
- Leucemia Mieloide Aguda não Especificadas
- Leucemia mieloide aguda com diferenciação mínima (M0).
- Leucemia mieloide aguda sem maturação (M1).
- Leucemia mieloide aguda com maturação (M2).
- Leucemia mielomonocítica aguda (M4).
- Leucemia monocítica aguda (M5).
- Leucemia eritróide aguda (M6).
- Leucemia megacarioblástica aguda (M7).
- Leucemia basofílica aguda.
- Leucemia mieloide aguda com fibrose.
- Sarcoma Mieloide ou Sarcoma Granulocítico ou Cloroma
- Proliferações Mieloides relacionadas com a Síndrome de Down
- Leucemias Agudas Indiferenciadas e Bifenotípica
Leucemia Linfoide Aguda (LLA)
- Hemograma completo (exame de sangue): a doença provoca aumento significativo dos glóbulos brancos, e também pode atingir os glóbulos vermelhos e as plaquetas.
- Mielograma
- Biópsia da medula
- O exame decitogenética, feito por meio de pequena amostra de sangue, irá analisar as alterações específicas das células, e assim determinar o subtipo da doença – ou seja, se é ou não uma leucemia aguda.
- Já aimunofenotipagem, por sua vez, feita também com uma amostra de sangue, irá verificar as características físicas das células – que geralmente são divididas em LLA tipo T e LLA tipo B (as células T e B são as afetadas).
- Caso o médico desconfie de um envolvimento do sistema nervoso central, ele também pode solicitar um estudo líquido da espinha (líquor). O aumento do baço e fígado, comum a alguns pacientes com LLA, devem ser analisados por meio de exames de imagens, como aultrassonografia.
- Para descobrir se o paciente apresenta o cromossomo Philadelphia, o médico poderá pedir o FISH (Hibridização Fluorescente in situ) e o PCR (reação em cadeia da polimerase).
Estadiamento
A leucemia linfocítica aguda por outro lado, normalmente não forma massas tumorais, mas geralmente afeta toda a medula óssea e, em muitos casos, pode se espalhar para outros órgãos, como o fígado, baço e gânglios linfáticos. Por conseguinte, a perspectiva para o paciente com leucemia linfoide aguda depende de outras informações, como subtipo de LLA, idade, e resultados de exames de laboratório.
Diferentes sistemas são utilizados para classificar a leucemia linfoide aguda em subtipos.
Classificação Francesa-Americana-Britânica (FAB)
Na década de 1970, um grupo de franceses, americanos e britânicos (FAB), especialistas em leucemia dividiram a leucemia linfoide aguda em três subtipos (L1, L2 e L3), baseados na maneira como suas células eram vistas ao microscópio após coloração de rotina. Este sistema foi substituído em função dos novos exames de laboratório que permitem uma classificação da leucemia linfoide aguda com mais precisão.
Classificação baseada no Imunofenótipo
Os exames de citogenética, citometria de fluxo, e outros exames de laboratório fornecem informações detalhadas sobre o subtipo de leucemia linfoide aguda e o prognóstico do paciente, o que permite classificar a leucemia linfoide aguda em grupos com base no imunofenótipo da leucemia, levando em conta o tipo de linfócitos (células B ou T) e a maturação das células leucêmicas. Os subtipos da leucemia linfoide aguda são:
- Provenientes dos tipos de linfócitos (células B ou células T).
- Maturidade das células leucêmicas.
Estes grupos têm substituído em grande parte a classificação FAB. Os subtipos de leucemia linfoide aguda são atualmente denominados como:
Leucemia Linfoide Aguda de Células B
- Leucemia linfoide aguda precoce B (10% dos casos).
- Leucemia linfoide aguda comum (50% dos casos).
- Leucemia linfoide aguda pré B (10% dos casos).
- Leucemia linfoide aguda B maduras, subtipo linfoma/leucemia de Burkitt (4% dos casos).
Leucemia Linfoide Aguda de Células T
- Leucemia linfoide aguda pré T (5% a 10% dos casos).
- Leucemia linfoide aguda T maduras (15% a 20% dos casos).
REFERÊNCIAS
- Instituto Oncoguia
- ABRALE – Associação Brasileira de Linfoma e Leucemia
7. Identificar as principais formas e fases do tratamento das leucemias e suas indicações.
Tratamento da Leucemia Linfoblástica Aguda
Depois que a condição das pacientes estiver estabilizada, o tratamento antileucêmico deve ser iniciado o mais depressa possível. O tratamento da LLA recém-diagnosticada pode ser dividido em três fases: indução da remissão, terapia pós-remissão e profilaxia do SNC.
- Indução da Remissão: O objetivo inicial do tratamento é induzir uma remissão completa, que é geralmente definida como sendo a redução dos blastos leucêmicos para níveis indetectáveis e a restauração da função medular normal. Uma variedade de esquemas quimioterápicos pode ser usada para induzir a remissão; todos incluem a vincristina e a prednisona, e a maioria deles acrescenta a L-asparaginase e/ou daunorrubicina administradas em um período de 3 a 4 semanas. Com tais esquemas, conseguimos uma remissão completa em 90% das crianças e 80% a 90% dos adultos (Tabela 189-2). Como a vincristina, a prednisona e a L-asparaginase são pouco tóxicas para os precursores de medula óssea, o paciente frequentemente entra em remissão completa depois de um período relativamente curto de mielossupressão. A incapacidade de atingir uma remissão completa se deve geralmente tanto à resistência das células leucêmicas às drogas usadas, quanto a uma infecção progressiva. Essas duas complicações ocorrem aproximadamente com igual frequência.
- Quimioterapia Pós-remissão: Se não houver uma continuidade no tratamento depois da indução da remissão completa, todos os casos poderão recidivar, a maioria depois de diversos meses. Esse fato demonstra a necessidade de prosseguir a quimioterapia após a remissão completa, que pode ser administrada em diversas associações, doses e esquemas. O termo quimioterapia para consolidação se refere em geral a ciclos curtos de quimioterápicos administrados com doses semelhantes às usadas para a indução inicial, e, assim, algumas vezes haverá necessidade de uma nova hospitalização. Em geral, tenta-se selecionar drogas diferentes para a consolidação daquelas usadas para induzir a remissão inicial. No caso da LLA, tais drogas compreendem o metotrexate em altas doses, a ciclofosfamida e a citarabina, entre outras. A manutenção implica a administração de quimioterapia em baixas doses numa base diária ou semanal, em ambulatório, por longos períodos de tempo. O esquema de manutenção mais frequentemente usado na LLA é o que associa 6-mercaptopurina diária e metotrexato semanalmente ou duas vezes por mês. Não se sabe a duração ótima para a quimioterapia de manutenção, mas, em geral, ela é administrada durante 2 a 3 anos. A quimioterapia ótima para a LLA exige consolidação e quimioterapia de manutenção.
- Profilaxia do Sistema Nervoso Central: A maioria dos agentes quimioterapêuticos que são administrados por via intravenosa ou oral não penetra bem no SNC, e se nenhuma forma de profilaxia do SNC for dada, pelo menos 35% dos adultos irão desenvolver leucemia do SNC. Com a profilaxia, a recidiva no SNC como um evento isolado ocorre em menos de 10% dos pacientes. No entanto, a quimioterapia sistêmica com altas doses de metotrexate (p. ex., 200 mg/m2 intravenosos durante 2 horas, seguidos por 800 mg/m2 durante 22 horas) e citarabina (p. ex., 3 g/m2 durante 2 horas, de 12 em 12 horas, em quatro doses) pode atingir níveis terapêuticos das drogas dentro do SNC. As alternativas são o metotrexate intratecal, o metotrexate intratecal associado a administração de radioterapia na dose de 2.400 cGy no crânio, ou 2.400 cGy no neuroeixo.
Tratamento da Leucemia Mieloide Aguda
- Indução da Remissão: O tratamento com uma combinação de uma antraciclina com citarabina (100 a 200 mg/m2/dia por 7 dias) leva a uma remissão completa em 60% a 80% dos pacientes portadores de LMA. Estudos clínicos prospectivos e randomizados demonstraram que a idarrubicina (10 a 12 mg/m2/dia por 3 dias) ou uma dose mais alta de daunorrubicina (60 a 90 mg/m2/dia por 3 dias) são superiores à dose convencional de daunorrubicina de 45 mg/m2/dia por 3 dias.3-5 Uma mielossupressão profunda sempre se segue quando esses agentes são utilizados em doses capazes de atingir a RC. A incapacidade de atingir uma RC é geralmente causada tanto pela resistência às drogas quanto pelas complicações fatais da mielossupressão.
- Quimioterapia Pós-remissão: Uma quimioterapia intensiva de consolidação com ciclos repetidos de daunorrubicina e citarabina em doses convencionais, citarabina em alta dose (1 a 3 g/m2/dia por 2 a 6 dias) ou outros agentes prolonga a duração média da remissão e aumenta as chances de sobrevida livre de doença em longo termo. Os melhores resultados descritos até agora com quimioterapia empregaram em geral ciclos repetidos de altas doses de citarabina. Ao contrário do preconizado para a LLA, o tratamento de manutenção com baixas doses depois do tratamento de consolidação intensivo apresenta benefícios limitados. Na LMA, a recidiva leucêmica ocorre com menor frequência no SNC, sendo observada apenas em aproximadamente 10% dos casos, na maioria das vezes nos pacientes com variantes M4 ou M5. Não existem evidências de que a profilaxia do SNC melhore a sobrevida livre de doença na LMA.
Escolha Inicial do Tratamento para a Leucemia Mieloide Crônica
As decisões de tratamento na LMC baseiam-se na fase da LMC, na idade do paciente, e na disponibilidade de um doador de célula-tronco. Para a LMC na fase crônica (>90% dos casos recém-diagnosticados), o mesilato de imatinibe, um inibidor seletivo de atividade tirosina quinase da proteína BCR-ABL, é a terapia de primeira linha para maioria dos pacientes. O transplante de células-tronco alogênico é considerado um tratamento de segunda linha eficaz para a LMC em fase crônica após falha do imatinibe. Terapias de segunda linha alternativas são mais potentes que os inibidores de tirosina quinase de segunda geração, como o dasatinibe (inibidor duplo das quinases de SRC-ABL) e o nilotinib (inibidor seletivo e mais potente de BCR-ABL). Os pacientes com LMC em fase acelerada ou blástica devem considerar o transplante de células-tronco alogênico como tratamento imediato definitivo; nesta situação, o uso de inibidores de tirosina quinase como uma medida de intervalo antes do transplante de células-tronco pode reduzir significativamente a carga de LMC e melhorar os resultados do transplante de células-tronco alogênicas.
- Mesilato de Imatinibe: Desde sua descoberta em 1999, o mesilato de imatinibe tornou-se a terapia padrão para LMC. O imatinibe é um derivado da 2-fenilaminopirimidina que se liga ao sítio canônico do trifosfato de adenosina (ATP) que reveste a fenda entre os lobos N e C do domínio de ABL com atividade de quinase, bloqueando a fosforilação da proteína substrato nos seus resíduos tirosina. O bloqueio da ligação do ATP à molécula ABL inativa sua atividade de quinase, porque esta não pode transferir o fosfato ao substrato. Pela inibição da fosforilação, o imatinibe previne a ativação de vias de transdução de sinal que induzem os processos de transformação leucêmica que causam a LMC (Fig. 190-4). O imatinibe bloqueia diversas tirosina quinases, incluindo p210BCR-ABL, p190 BCRABL, v-ABL, c-ABL, c-Kit e o receptor de PDGF.
Referência: Goldman Cecil Medicina, Capítulo 189: Leucemias agudas| Lee Goldman, MD and Andrew I. Schafer, MD,https://www.evolution.com.br/epubreader/goldman-cecil-medicina-24ed
8. Discutir a importância da equipe multidisciplinar para acompanhamento de pacientes e seus familiares.
- Serviço Social Hospitalar
Abrange um conjunto de ações, que norteado pelos princípios da profissão, busca garantir por intermédio dos recursos sociais disponíveis, viabilizar condições para a adesão ao tratamento médico do pacientes e família em um processo de promoção humana.
- Assistente Social na Equipe
O Assistente Social é o profissional que observa, analisa e intervém direta e indiretamente nas mais diversificadas situações socioeconômicas e emocionais apresentadas pelos pacientes e seus familiares.
E juntamente com a equipe multiprofissional, está receptivo a todas as ações profissionais que contribuem para integração do paciente à sociedade, otimizando o tratamento, de tal forma que a doença passe a ser encarada como uma “circunstância”, a qual não impedirá paciente e família à ascenderem êxitos educacionais e/ou profissionais.
O trabalho em equipe é fundamental para uma assistência integral ao paciente e a família. A equipe deverá atender este paciente da forma mais abrangente possível e com base nas necessidades do mesmo e de sua família.
Este acolhimento deve ser gradual e contínuo, estabelecendo desta forma, uma relação mais próxima com a equipe, sempre buscando uma maior qualidade no atendimento.
- Conversando sobre o diagnóstico
Quando o paciente é atendido pela equipe e já soube de seu diagnóstico, ele recebe o acolhimento. O assistente social auxilia na interpretação da situação médica e sobre o tratamento proposto.
A equipe multidisciplinar deve sempre ter como objetivo promover uma ação educativa, buscando um processo reflexivo nos pacientes, para que possam participar do processo de tratamento e (ou) cura.
Referências: ABRALE – Associação Brasileira de Linfoma e Leucemia
9. Identificar o papel das entidades de apoio ao paciente com câncer e familiares.
Existem ONGS como o GRAAC, GAPC, CASA HOPE, ABRAPEC etc. que dão todo o suporte psicológico, financeiro e médico que os pacientes oncológicos precisam, visto que muitas vezes eles não têm condições de bancar e arcar tais custos.
- Equipe de psicologia da ABRALE
O Departamento de Psicologia da ABRALE oferece diferentes formas de apoio psicológico, que têm por objetivo a saúde global, o bem-estar das pessoas e o reforço aos resultados dos tratamentos médicos.
Em uma entrevista individual, nossos psicólogos especializados buscam compreender as expectativas e necessidades de cada pessoa a fim de nortear o foco do trabalho terapêutico, se for necessário. É a partir da avaliação psicológica que se estabelece o plano de cuidados, que pode consistir em psicoterapia individual, em orientações específicas ou em encaminhamentos para programas internos ou externos.
Baseada nos dados levantados durante a avaliação psicológica, o psicólogo da nossa equipe define juntamente com o paciente um foco a ser trabalhado, o objetivo do processo terapêutico. Também é planejado o número de encontros terapêuticos, entre 8 e 12 sessões, que acontecem uma vez por semana. Em casos especiais, o número de semanas e a frequência dos encontros pode mudar. Ao final do processo, são verificados os resultados alcançados. Os recursos utilizados são diversos e incluem técnicas de imagética, hipnose e relaxamento, entre outras.
O GRAACC é uma instituição sem fins lucrativos, criada para garantir a crianças e adolescentes com câncer, dentro do mais avançado padrão científico, o direito de alcançar todas as chances de cura com qualidade de vida. O hospital do GRAACC possui cerca de 3.000 crianças atendidas anualmente, entre sessões de quimioterapia, consultas, procedimentos ambulatoriais, cirurgias, transplantes de medula óssea e outros. Além de diagnosticar e tratar o câncer infantil, o GRAACC atua no desenvolvimento do ensino e pesquisa.
O GRAACC nasceu em 1991, graças à iniciativa do Dr. Sérgio Petrilli, chefe do setor de Oncologia do Departamento de Pediatria da Escola Paulista de Medicina, o engenheiro voluntário Jacinto Antonio Guidolin e Sra. Léa Della Casa Mingione, voluntária do Hospital do Câncer.
O primeiro passo foi transferir o Setor de Oncologia Pediátrica do Hospital São Paulo para uma casa, que ficou conhecida como a “casinha”. Os pequenos pacientes eram atendidos nesse local, dentro do conceito de hospital-dia, onde recebiam atendimento médico e assistencial e voltavam para as suas casas.
Fundamentado na parceria universidade/empresa/comunidade, o GRAACC despertou em empresas e instituições de larga visão social a confiança e o interesse em participar da construção do Instituto de Oncologia Pediátrica – IOP/GRAACC/UNIFESP, o hospital do GRAACC.
O hospital é gerenciado e administrado pelo GRAACC e a assistência médica, o ensino e a pesquisa são conduzidos em convênio com a Universidade Federal de São Paulo (UNIFESP/EPM).
Referências: ABRALE – Associação Brasileira de Linfoma e Leucemia; INCA; GRAAC.









Comentários